

Article Sidebar
Downloads
Introduction
Intellectual disability (ID) is a neurodevelopmental disorder characterized by the impairment of skills manifested during the developmental period, which contribute to the overall level of intelligence. The prevalence is estimated at 1% [1].
ID can present as nonsyndromic (ID is the sole clinical feature) or syndromic (with additional symptoms), although this distinction is not always obvious [2]. Due to the high male-to-female ratio, X-linked ID has been extensively examined. This has led to the discovery of more than 80 genes associated with ID on the X chromosome (of which 30 cause nonsyndromic ID). Since the X chromosome represents only about 5% of the human genome there is increasing evidence that there are many autosomal–recessive loci involved in ID [3]. It is estimated that autosomal-recessive ID accounts for 40% of all individuals with nonsyndromic forms, and is extremely prevalent in consanguineous populations [4]. Recently, over 300 genes have been identified to play a role in autosomal-recessive forms of ID. More than 97% of them have a role in a recessive disorder that includes ID as one of their symptoms, only a few genes are identified as giving ID as the main phenotype [5]. One of these is the TRAPPC9 gene. Homozygous mutations have been described in several families and are linked with moderate-to-severe ID associated with typical facial appearance, obesity, early onset hypotonia and brain anomalies [6]. Here, we describe three siblings in whom a homozygous nonsense mutation of TRAPPC9 was identified using exome sequencing analysis. Previously described phenotypical features were found in combination with progressive encephalopathy and destructive white matter lesions, suggesting a neurodegenerative disease.
Patients
Three siblings (two boys and one girl) were referred to the paediatric neurologist because of neurodevelopmental delay. Their parents were healthy consanguineous (cousins) of Algerian ethnicity.
The two brothers were followed for many years in our center. They both were born at term after an uneventful pregnancy. During the first year of life, no pronounced abnormalities were found. It was only in their second year that both boys presented with similar symptoms of neurodevelopmental delay and epilepsy. Phenotypically some peculiar similarities were found, including pronounced microcephaly, flat occiput, brittle hair and syndactyly of the second and third toe. (Figure 1)
Figure 1.Phenotype of the three siblings.
In the following years, it became clear that their developmental delay was severe. They presented with general hypotonia and were never able to walk unsupported. Intellectually they were severely disabled with an almost absent speech. Both developed autistic traits with prominent behavioral problems and stereotypic movements, which needed treatment by different antipsychotic drugs. Partial epileptic seizures were seen during childhood and were successfully treated with sodium valproate.
Extensive etiological work-up was performed in both. Electrophysiological tests consisted of an electroencephalogram (EEG), Brainstem Evoked Response Audiometry (BERA) and somatosensory evoked potentials (SSEP). EEG showed low voltage and little variability. BERA and SSEP were normal. A complete metabolic profile using blood, urine and cerebrospinal fluid or muscle and skin biopsies did not identify any abnormalities. Genetic work-up showed a normal male karyotype, no 22q11 deletion and the absence of fragile X-syndrome.
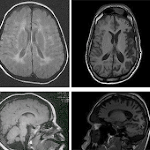
Repetitive magnetic resonance imaging (MRI) was performed in both. As shown in Figure 2, the MRI of the oldest boy demonstrated colpocephaly, mega cisterna magna, partial agenesis of the corpus callosum, cerebellar medio-inferior hypoplasia, thin white matter, abnormal signal intensity in the occipito-temporal white matter, normal cortical morphology and no brainstem involvement.
Figure 2.MRI of the oldest boy.
In the youngest boy, MRI demonstrated mega cisterna magna, hypoplasia of the corpus callosum, cerebellar inferior hypoplasia, abnormal bilateral postero-parietal and occipital white matter, white matter hypoplasia, mild ventricular dilatation, no basal ganglia involvement and a normal brainstem. In both the repeated MRI’s showed progressive ventricular dilatation with decrease of white matter and cortical atrophy (Figure 3).
Figure 3.MRI of the youngest boy.
The first 2 images show the first MRI, the second 2 images show the progression.
After giving birth to two healthy sisters, a third sibling girl was born with similar symptoms. She was born at term after an uneventful pregnancy. She was first examined at one year of age. The mother reported slight hypotonia during the first months of life, with the inability to sit or crawl. Phenotypically she demonstrated the same distinctive craniofacial appearance with microcephaly and flat occiput (Figure 1). In the following years, the hypotonia continued with inability to walk unsupported. She was also intellectually disabled with almost absent speech and demonstrated autistic traits with stereotypic movements which needed treatment by antipsychotic drugs. No epileptic activity was seen. MRI showed similarities with those of her brothers with hypoplasia of the corpus callosum, a delayed myelination and abnormal bilateral white matter (Figure 4).
Figure 4.MRI of the sister.
Since first genetic and metabolic work-up was negative, a whole exome sequencing was performed according to standard procedures for one of the sibs. This revealed a homozygous c.1708C>T, p. Arg570 mutation in the TRAPPC9 gene. The same mutation was also found by Sanger sequencing in both brothers, and the parents were heterozygous carriers of the mutation. The two unaffected sisters were not carrying the mutation. No other homozygous or compound heterozygous mutation could be detected that might explain the phenotype of the patients. (Figure 5)
Figure 5.Family pedigree
Discussion
To date, autosomal recessive nonsyndromic ID caused by homozygous TRAPPC9 mutations have been reported in eight families [2, 3, 4][6, 7, 8]. In all cases this resulted in loss-offunction mutations, which resulted in the almost complete absence of TRAPPC9 protein by western blot analysis [3,7]. The consequence of c.1708C>T mutation on protein expression was evaluated in 2009 by Philippe et al and showed complete absence of the TRAPPC9 protein in skin fibroblasts [7].
In Table 1 the clinical signs associated with TRAPPC9 mutation are summarized. Several of them are variably present in our patients. Unlike the previously reported happy disposition as a psychiatric symptom, we see that in our patients behavioral manifestations were more severe with the need for antipsychotic drugs. The ID in our patients was extensive with the almost complete absence of spoken language and the inability to walk alone.
| CATEGORY | SUBCATEGORY | FEATURES |
|---|---|---|
| Growth | Weight | • Truncal obesity |
| Head and neck | Head | • Microcephaly |
| Face | • Short, smooth philtrum | |
| Eyes | • Wide nasal bridge | |
| • Hypertelorism | ||
| • Hypotelorism | ||
| • Hypoplastic supraorbital ridges | ||
| • Straight eyebrows | ||
| • Downturned mouth | ||
| • Clefth lip | ||
| • Prominent central incisors | ||
| • Short neck | ||
| Skeletal | Hands | • Long, thin fingers |
| Neurologic | Central nervous system | • Mental retardation, moderate to severe |
| • Language development limited to a few words | ||
| • Seizures | ||
| • Delayed walking | ||
| • Hypotonia | ||
| • Absent continence | ||
| • Thin corpus callosum | ||
| • Decreased volume of the cerebral white matter | ||
| • White matter abnormalities | ||
| • Hypoplasia of the inferior cerebellar vermis | ||
| Behavioral | • Happy disposition | |
| Psychiatric | • Hyperactivity | |
| Manifestations | • Stereotypic movements | |
| Miscellaneous | • Onset in infancy |
The MRI abnormalities as noted above are consistent with earlier reports. Since the two brothers were followed for more than two decades, it was possible to obtain several consecutive MRI’s. This revealed progressive white matter lesions. In combination with the presence of a normal development during the first year, the development of progressive microcephaly and the appearance and even worsening of symptoms during the following years suggest that TRAPPC9 mutation is not only a cause of ID, but should be considered as a form of a neurodegenerative disease. An explanation can be found in the function of the TRAPPC9 protein, which is involved in the nuclear factor-kappaB (NF-κB) signaling pathway which on its turn plays a role in a number of cellular processes such as immunity, cellular proliferation, apoptosis and development. Previous reports stated the role of NF-κB in different neurodegenerative diseases such as Alzheimer’s disease, Parkinson’s disease, Huntington’s disease and AIDS dementia [9]. The precise role of NF-κB in ID remains uncertain. Cognitive dysfunction is considered as the presence of defects in synapse formation and plasticity. NF-κB seems to play an important role in synaptic plasticity and long-term-memory formation [10]. Since recent data suggest an active role of astrocytes and oligodendrocytes in synaptic transmission, impaired expression in glial cells may also play a role. At last, NF-κB has been shown to establish the axon myelination of peripheral neurons by Schwann cells, it is also hypothesized that it plays the same role in the differentiation of oligodendrocytes [7].
In conclusion we can state that TRAPPC9 mutation should be considered in children with a typical phenotype and progressive neurodevelopmental delay. Long term follow- up, repetitive evaluation, consecutive MRI and further examination of the exact function of TRAPPC9 protein in the central nervous system is needed to determine if TRAPPC9 mutation should be classified as a neurodegenerative disease.
Acknowledgments
None.
Competing interests
The authors declare that they have no competing interests.
Author contributions
All authors have made substantial contributions to conception and design, acquisition of data, analysis and interpretation of data, have been involved in drafting the manuscript and revising it critically for important intellectual content and have given final approval of the version to be published.
This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/ licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly credited. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
References
- Maulik PK, Mascarenhas MN, Mathers CD, Dua T, Saxena S. Prevalence of Intellectual Disability: A Meta-analysis of Population-based Studies.. Research in Developmental Disabilities. 2011; 32:419-36. DOI | PubMed
- Mir A, Kaufman L, Noor A, Motazacker MM, Jamil T, Azam M, Kahrizi K, Rafiq MA, Weksberg R, Nasr T, Naeem F, Tzschach A, Kuss AW, Ishak GE, Doherty D, Ropers HH, Barkovich AJ, Najmabadi H, Ayub M, Vincent JB. Identification of Mutations in TRAPPC9, wich Encodes the NIK- and IKK-β-Binding Protein, in Nonsyndomic Autosomal-Recessive Mental Retardation.. The American Journal of Human Genetics. 2009; 85:909-15. DOI | PubMed
- Mochida GH, Mahajnah M, Hill AD, Basel-Vanagaite L, Gleason D, Hill RS, Bodell A, Crosier M, Straussberg R, Walsh CA. A Truncating Mutation of TRAPP9C is Associated with Autosomal-Recessive Intellectual Disability and Postnatal Mircocephaly.. The American Journal of Human Genetics. 2009; 85:897-902. DOI | PubMed
- Kabar N, Goebel I, Daud S, Nürnberg G, Agha N, Ahmad A, Nürnberg P, Kubisch C, Ahmad J, Brock G. A Homozygous Splice Site Mutation in TRAPP9C Causes Intellectual Disability and Microcephaly.. European Journal of Medical Genetics. 2012; 55:727-31. DOI | PubMed
- Vissers L, Gilissen C, Veltman J. Genetic Studies in Intellectual Disability and Related Disorders.. Nature Genetics. 2016; 17:9-18. DOI | PubMed
- Marangi G, Leuzzi V, Manti F, Lattante S, Orteschi D, Pecile V, Neri G, Zollino M. TRAPP9C-related Autosomal Recessive Intellectual Disability: Report of a New Mutation and Clinical Phenotype.. European Journal of Human Genetics. 2013; 21:229-32. DOI | PubMed
- Philippe O, Rio M, Carioux A, Plaza JM, Guigue P, Molinari F, Boddaert N, Bole-Feysot C, Nitschke P, Smahi A, Munnich A, Colleaux L. Combination of Linkage Mapping and Microarray-Expression Analysis Identifies NF- κB Signaling Defect as a Cause of Autosoma-Recessive Mental Retardation.. The American Journal of Human Genetics. 2009; 85:903-8. DOI | PubMed
- Abou Jamra R, Wohlfart S, Zweier M, Uebe S, Priebe L, Ekici A, Giesebrecht S, Abboud A, Al Khateeb MA, Fakher M, Hamdan S, Ismael A, Muhammad S, Nöthen MM, Schumacher J, Reis A. Homozygosity mapping in 64 Syrian Consanguineous Families with Non-specific Intellectual Disability Reveals 11 Novel Loci and High Heterogeneity.. European Journal of Human Genetics. 2011; 19:1161-6. DOI | PubMed
- Hu WH, Pendergast JS, Mo XM, Brambilla R, Bracchi-Ricard V, Li F, Walters WM, Blits B, He L, Schaal SM, Bethea JR. NIBP, a novel NIK and IKK(beta)-binding protein that enhances NF-(kappa)B activation. Journal of Biological Chemistry. 2005; 280:29223-41. DOI | PubMed
- Romano A, Freudenthal R, Merlo E, Routtenberg A. Evolutionarily-conserved Role of the NF-kappaB Transcription Factor in Neural Plasticity and Memory.. European Journal of Neuroscience. 2006; 24:1507-16. DOI | PubMed
Main Article Content
Abstract
Intellectual disability is an important health issue, with a prevalence estimated at 1%. Autosomal genes are increasingly identified as an important cause, although still few of them are known. Most cases are of recessive origin and are found in large consanguineous families in the Middle East. Mutations in the TRAPPC9 gene have been reported in different families and are associated with a nonsyndromic form of intellectual disability, although phenotypic abnormalities such as a specific facial appearance, obesity, hypotonia and consistent brain abnormalities were linked to these mutations. Here, we describe three siblings of consanguineous Algerian parents with a homozygous c.1708C>T, p. Arg570* mutation in the TRAPPC9 gene. Initially a relatively normal development was seen until the age of 12-18 months, with from then on a progressive degradation of their mental and motor state and the presence of convulsions in two of them. This, in combination with progressive white matter lesions seen on repetitive magnetic resonance imaging, suggests that this TRAPPC9 mutation should not only be considered as a form of intellectual disability, but also as a neurodegenerative disease.
Article Details

This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License.