

Article Sidebar
Main Article Content
Abstract
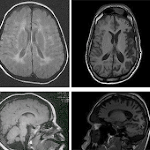
Intellectual disability is an important health issue, with a prevalence estimated at 1%. Autosomal genes are increasingly identified as an important cause, although still few of them are known. Most cases are of recessive origin and are found in large consanguineous families in the Middle East. Mutations in the TRAPPC9 gene have been reported in different families and are associated with a nonsyndromic form of intellectual disability, although phenotypic abnormalities such as a specific facial appearance, obesity, hypotonia and consistent brain abnormalities were linked to these mutations. Here, we describe three siblings of consanguineous Algerian parents with a homozygous c.1708C>T, p. Arg570* mutation in the TRAPPC9 gene. Initially a relatively normal development was seen until the age of 12-18 months, with from then on a progressive degradation of their mental and motor state and the presence of convulsions in two of them. This, in combination with progressive white matter lesions seen on repetitive magnetic resonance imaging, suggests that this TRAPPC9 mutation should not only be considered as a form of intellectual disability, but also as a neurodegenerative disease.
Article Details
Copyright (c) 2018 Helen Franckx

This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License.
Authors who publish with this journal agree to the following terms:
Authors retain copyright and grant the journal right of first publication with the work simultaneously licensed under a Creative Commons Attribution License that allows others to share the work with an acknowledgement of the work's authorship and initial publication in this journal.
Authors are able to enter into separate, additional contractual arrangements for the non-exclusive distribution of the journal's published version of the work (e.g., post it to an institutional repository or publish it in a book), with an acknowledgement of its initial publication in this journal.
Authors are permitted and encouraged to post their work online (e.g., in institutional repositories or on their website) prior to and during the submission process, as it can lead to productive exchanges, as well as earlier and greater citation of published work (See The Effect of Open Access).