

Article Sidebar
Main Article Content
Abstract
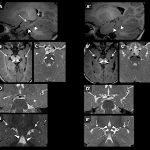
Hypochondroplasia is a disorder caused by FGFR3 mutations, commonly featuring short limbs and stature. Thanatophoric dysplasia, achondroplasia and Muenke syndrome are genetically related conditions with skeletal dysplasia and temporal lobe dysgenesis. We present a 6-month old boy with hypochondroplasia and epilepsy. His brain MRI revealed bilateral temporal lobe dysplasia, with redundant sulci and a rotated hippocampus. The neuroimaging findings of our case provide direct insight in the pathogenesis of the cerebral dysgenesis in FGFR3-related disorders. In addition, through this case we review the neurological spectrum of FGFR3 mutations.
Article Details
Copyright (c) 2018 Jurriaan M Peters

This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License.
Authors who publish with this journal agree to the following terms:
Authors retain copyright and grant the journal right of first publication with the work simultaneously licensed under a Creative Commons Attribution License that allows others to share the work with an acknowledgement of the work's authorship and initial publication in this journal.
Authors are able to enter into separate, additional contractual arrangements for the non-exclusive distribution of the journal's published version of the work (e.g., post it to an institutional repository or publish it in a book), with an acknowledgement of its initial publication in this journal.
Authors are permitted and encouraged to post their work online (e.g., in institutional repositories or on their website) prior to and during the submission process, as it can lead to productive exchanges, as well as earlier and greater citation of published work (See The Effect of Open Access).